Computer Graphics and DNA Sequencing
Michael J. Potel
Taligent
Pundits have called the 20th century the century of physics but predict the 21st will be the century of biology. A key development driving this profound change is the biotechnology of DNA (deoxyribonucleic acid) sequencing and genetic analysis. A combination of sophisticated biochemical procedures, laboratory instruments, and computer methods have made it possible to determine the genetic code sequences present in DNA, the blueprint for life itself. The result has been unprecedented progress in detecting and treating diseases, improving organic products like food, chemicals, and drugs, determining identities and hereditary relationships, and harnessing the power of living systems (see the sidebar "Application areas").
DNA sequencing is a means for determining the exact series of genetic codes in the DNA of cell nuclei. The DNA double helix encases a long ladder-step sequence of base pairs made of the nucleotides adenine, thymine, guanine, and cytosine (A, T, G, and C). These base pairs encode the amino acids used when the DNA acts as a template for building proteins. Proteins carry out all the key chemical operations in the cell, controlling everything from eye color to food digestion, fighting disease, and building muscles, organs, and tissues. Many proteins act as enzymes, which in turn govern the creation and use of all the other molecules in the cell. Knowing a cell’s DNA sequence thus amounts to having a specification for all the components that implement a cell’s function and behavior (see the sidebar "Sizing the problem").
The role of interactive graphics systems
Today’s biotechnology revolution would not have happened without computers. Powerful interactive computer graphics applications lie at the core of DNA sequencing work, which represents the most widespread and successful use of computers in the biological sciences. Complex biochemical processes lack counterparts to the laws of physics and the rigor of mathematics driving the physical sciences. But the comparatively "digital" DNA sequencing problem has benefited greatly from algorithmic processes, large-scale combinatorics, and discrete mathematics.Much of the computing for DNA sequencing research was developed on Unix workstations because of their processing power, big address spaces, and database and storage capacities. As DNA sequencing moved into industrial and clinical domains, additional requirements such as ease of use, systems manageability, and affordability emerged. Fortunately, at the same time much more powerful and affordable personal computers, with better PC operating systems, networking, and databases, also appeared. As DNA sequencing gains commercial acceptance, the most successful product lines are now dominated by Macintosh and Windows-based PCs, with Unix used for servers and databases.
The computer graphics of DNA sequencing has a different character than the graphics of image rendering. It entails highly interactive systems that allow researchers to organize and analyze the massive data sets inherent in the DNA sequencing process. These systems use complex graphs, multiple synchronized views and representations, rapid navigation and measurement, sophisticated pattern matching and searching, and large databases and networking. The applications are built as tools for scientific investigative work, not to produce graphics images. Nonetheless, building the computer programs used in DNA sequencing involves many interesting graphics problems.
DNA sequencing basics
DNA sequencing has four key aspects:
- Fragment sequencing—breaking DNA molecules into segments of a few hundred bases and determining their base pair sequences by clever analytic methods.
- Sequence assembly—comparing multiple fragment sequences for overlaps and piecing them together into gene-sized sequences.
- Database processing—storing, searching, comparing, and integrating sequences with those from other experiments and laboratories.
- Genotyping and linkage analysis—relating sequence data to useful biological information by mapping traits inherited by family members.
Fragment sequencing
DNA sequencing depends on breaking up the DNA strand into smaller subsets for which base pair sequences can be determined. In addition, the methodology used to sequence a given DNA fragment requires many copies of the fragment in question. A key biochemical procedure called the polymerase chain reaction, or PCR, meets both needs.
Kary Mullis invented PCR in 1983 while at Cetus, a genetic engineering company in Emeryville, California. The work earned him a Nobel prize. (Mullis has since gained additional notoriety for his involvement in last year’s O.J. Simpson trial.) PCR permits rapid, large-scale replication of a given section of a DNA molecule, employing machines that use fast cycling of hot and cold and special bacteria found in hot springs and geysers.
The DNA fragment for replication is selected between two DNA markers called primers that match unique regions a few dozen bases long in the DNA. Hundreds of primers for interesting areas of DNA are available commercially from biotechnology companies, as are machines that let researchers create their own primers for any desired ATGC sequence.
|
|
|
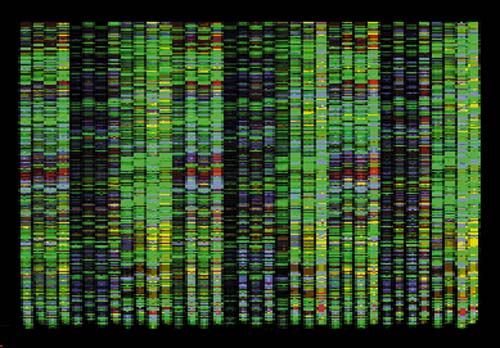
The basic DNA sequencing technique itself was invented in 1975 by Frederick Sanger of Cambridge University, for which he received a Nobel prize. A set of four special fluorescent markers are attached to the A, T, G, and C nucleotides, each responding with a different wavelength of light, typically red, yellow, green, and blue. Each labeled nucleotide is also modified so that it will terminate DNA replication once it attaches at one end of a growing strand. Adding the four fluorescent markers into a PCR process results in copies of the DNA fragment truncated at every possible base position along its length, with a fluorescent label identifying the terminating nucleotide. The sample is then run through an electrophoresis gel, which sorts fragments by size, resulting in a defining fluorescent marker for each nucleotide position (see Figure 1).
Computerized imaging techniques are indispensable in analyzing such gels. The gels are scanned with a laser optical system, resulting in an electropherogram—four overlapping graphs with the trace peaks indicating the color at each position (see Figure 2). Programs for base calling analyze these graphs to determine the desired ATGC sequence. But rarely does it work this easily. The fluorescent labeling is not so precise as to prevent overlap between the four colors. Also, the longer the fragment, the less precise the resolution and position of the peaks (bottom row of Figure 2). Filtering operations can improve the ability to resolve peaks. Another problem is that gels frequently stretch or distort, requiring "lane tracking" techniques to linearize them. Peak intensities and lane measurements are calibrated by running known standards in parallel on the gel. Most experiments involve multiple gels, so alignment and calibration of multiple data sets is also essential. Despite powerful base-calling algorithms, a fully automated process is not assured, so researchers need to examine large numbers of electropherograms interactively to guide the process, resolve conflicts, fix errors, and assess confidence levels.
The acknowledged leader in DNA sequencing systems is Perkin-Elmer’s Applied Biosystems Division in Foster City, California (http://www.perkin-elmer.com/ab/index.htm) with competing systems from Swedish company Pharmacia (http://www.biotech.pharmacia.se) and Li-Cor in Lincoln, Nebraska (http://www.licor.com/bio/biohome.htm).
Perkin-Elmer has specialized in systems for PCR-based DNA sequencing and assembly, including automated laboratory instruments and computer analysis programs, as well as biological reagents used in preparation and processing. They also make DNA synthesizers, which take ATGC sequences as input and create corresponding DNA strands for primers or for probes to test for specific sequences.
|
|
|
In the future, computer technology may provide even more innovative ways to perform DNA sequencing. A small company, Affymetrix in Santa Clara, California, is developing the GeneChip, a proprietary VLSI array able to read bases directly from DNA molecules. The idea is to spread DNA fragments over a VLSI array and detect the minute differences between the nucleotides at each position. This technology might some day surpass the effectiveness of fluorescence techniques.
Sequence assembly
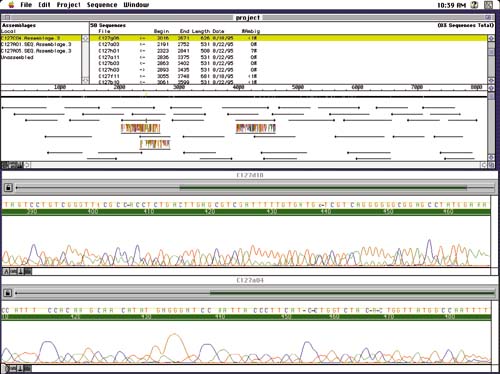
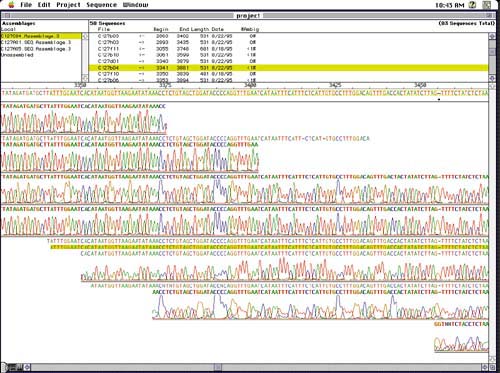
The next problem is to take many overlapping DNA fragment sequences a few hundred bases long and assemble them into longer segments of interest, such as genes spanning thousands of bases. Thousands of fragments must be sequenced and pieced together like a long jigsaw puzzle by matching up random overlapping ends, as in Figure 3. Interactive programs help shift and align multiple sequences, and correlations and other statistical inferences are used to reconstruct the overall sequence. A host of pattern matching techniques, plots and measures, and visual navigation tools assist this process.
Matching all possible overlaps of thousands of fragment sequences is hard enough. Complicating this problem, fragment sequences may have base calling errors. Sometimes the DNA replication process itself substitutes incorrect nucleotides or causes a base to be omitted or inserted. Sequence assembly must allow for these errors, greatly increasing the computation involved.
Another complication arises from the double-stranded nature of DNA. For every sequence in half a DNA strand, a genetic mirror sequence occurs in the other half, matching A, T, G, and C with the complementary base pair nucleotides T, A, C, and G, respectively, and running in the opposite direction. Each fragment sequence must also be considered in this alternate form. Assembly programs display arrows running left and right as they match different fragments in different directions (top section of Figure 2).
While some sequences are relatively unique, making it fairly easy to find and match them up, DNA also contains many "junk" sequences, with long stretches of repetitions, duplications, reversals, and even bits of foreign DNA from viruses. Thus, some subsequences could fit in multiple positions and orders. These relatively common regions of ambiguity necessitate computer-assisted analysis of large numbers of fragments to build up accurate sequences for whole genes.
Database processing
Many laboratories study particular DNA areas, such as those related to disease or with commercial implications. Certain organisms also have widespread research interest, such as E. coli (bacteria), yeast, Drosophila (fruit flies), or laboratory mice, and there are active efforts to sequence their entire genomes. The Human Genome Project—the ultimate such undertaking—has the goal of sequencing the entire three billion base pairs of the human species.
In these cases, sharing sequences benefits all concerned, though a few commercial projects keep their sequences confidential. Credibility is built when others can replicate work. Also, because of the great similarities among the DNA of all species, sequences from one species can provide valuable starting points and cross-checks for sequencing in another. An effort the size of the Human Genome Project can only be accomplished through the combined efforts of hundreds of laboratories world-wide.
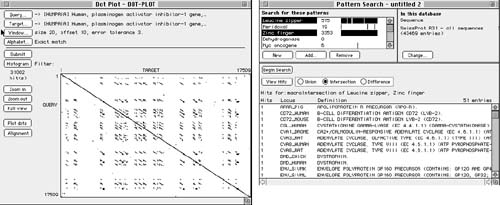
To these ends, organizations maintain large databases of DNA sequences, in many cases globally accessible on the Internet (for example, the On-line Mendelian Inheritance in Man database at http://www3.ncbi.nlm.nih.gov/omim, the Genome Sequence Database at http://www.ncgr.org/gsdb, and the Genome Database at http://gdbwww.gdb.org). Special interactive tools facilitate searching, matching, and comparison with known sequences. Programs such as those shown in Figure 4 let researchers enter regular expressions for sequences as templates for DNA pattern matching. These programs can search multiple databases and generate potentially large diagrams and tables showing all matching or near-matching areas of interest to facilitate gene identification and characterization.
With very large databases, searching speeds become a major factor. For high-speed searches, Perkin-Elmer has developed systems based on the Fast Data Finder chip originally developed by TRW for intelligence work and licensed in 1990. Each chip has a systolic array of 24 text-matching processors. A plug-in board holds 30 chips, a system contains up to five boards, and any number of systems is possible. These devices greatly facilitate comparisons with known sequences using different parameters and scoring matrices, supporting special capabilities for inexact matching and the other variants inherent in DNA sequence searching.
Genotyping and linkage analysis
DNA sequencing by itself only yields abstract data in the form of nucleotide codes. To use this data, researchers must relate it to biologically relevant properties and individual traits. The ultimate goal is to identify along the whole DNA genome not just the nucleotide sequence but also the traits and conditions controlled by particular genes and the proteins they encode.
Even before DNA sequencing, researchers could determine approximate locations on specific chromosomes of particular traits, using a technique known as linkage analysis. Linkage analysis takes advantage of how offspring inherit traits from their parents. Sexually reproducing organisms have two copies of each chromosome, one from each of their parents’ pairs. By examining where different traits show up in a family tree, we can identify which traits are associated with genes on the same chromosome, since they tend to be inherited together.
Actually, things are often more complex. During reproduction, chromosomes can cross over with the other member of a chromosome pair, forming hybrids. In this process, genes lying closer together on the same chromosome are less likely to become separated than ones further apart. With enough family members to evaluate and the right traits expressed, researchers can figure out not just which genes occur on what chromosomes but also what genes are adjacent on the same chromosome. Over the years, linkage maps for many traits have been worked out in humans and other species (visit the Cooperative Human Linkage Center at http://www.chlc.org or the site maintained by Jurg Ott at Rockefeller University, http://linkage.rockefeller.edu).
An intimate relationship exists between linkage analysis and DNA sequencing. Because linkage mapping can identify which genes lie near others, DNA sequencing of those genes can use primers based on sequences in nearby genes to which they are linked. Conversely, using techniques similar to DNA sequencing, but at a coarser granularity, permits measuring the relative positions and sizes of gene fragments. Given enough interplay between these techniques, researchers can determine the sequences for genes corresponding to specific traits, as well as overall physical maps locating identifiable DNA fragments in the chromosomes.
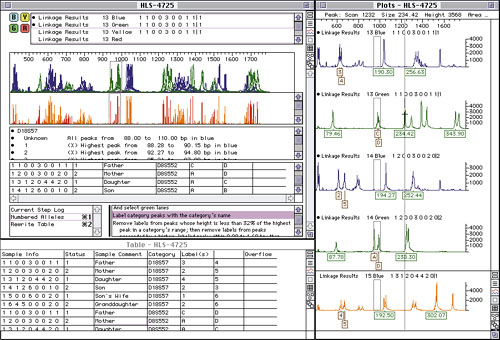
The techniques—and hence the computer programs, instruments, and reagents—for linkage analysis in many ways resemble DNA sequencing. Any identifiable location in the DNA is called a marker (or locus). Markers that vary and discriminate among individuals rouse the greatest interest, with the different possible values known as alleles. The identification and study of markers and their alleles is known as genotyping. PCR can amplify specific markers much as in sequencing, producing DNA fragments tagged with fluorescent labels. Electrophoresis gels are run and analyzed as before, producing graphs such as Figure 5. Analyzing the traces for the same markers in parents and their offspring and correlating these with inherited traits makes it possible to identify which alleles correspond to which traits.
This process has its share of difficulties in practice. Alignment and calibration between individuals is crucial. Important peaks are often adjacent and overlapping, requiring curve fitting and other statistical techniques to identify them. In addition, the PCR process by its nature tends to generate multiple false peaks and "echoes," requiring special heuristics and interactive techniques to sort them out. In the course of genotyping, researchers build and analyze large tables of traits and markers. Databases again prove invaluable in organizing these data and facilitating analyses.
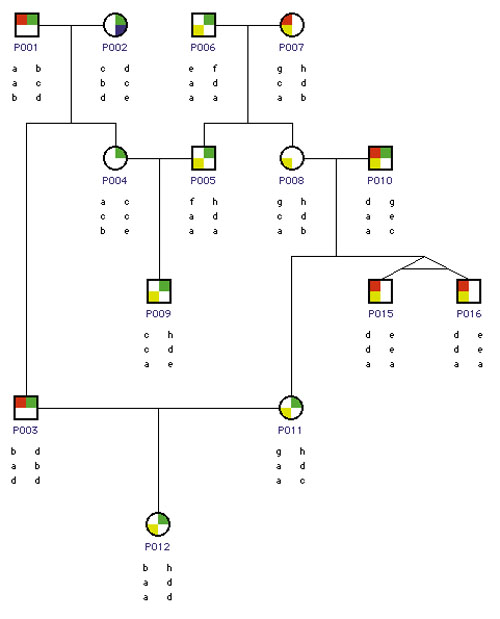
Diagramming traits and inherited relationships in complex family groups is another important capability. Pedigree drawing programs such as that illustrated in Figure 6 portray family trees and associated traits and alleles. A tree structure shows parents and offspring, and also represents special relationships such as identical twins. Different symbols indicate the sex and traits of individuals. Under each individual researchers will often plot alleles arranged in linkage order for both inherited chromosomes.
Drawing pedigree diagrams can be a hard graphical problem. Simple family relationships and small pedigrees are relatively straightforward. But complex relationships like multiple marriages, inbreeding, incest, and cross-generational unions prove much more difficult. Such relationships are common in animal experiments and breeding, and sometimes occur in humans in cases like the royal families of Europe. The resulting loops and cycles make it difficult to maintain desired adjacencies and draw pedigree diagrams without overlapping lines. Interesting graphical algorithms have been developed to determine optimal layout of generations, orderings within generations, and duplication of individuals as needed to maximize the comprehensibility of these diagrams. Examination of these graphs is often the key to understanding genetically determined traits and mapping them back onto specific DNA sequences and, by implication, the proteins that govern them.
Summary
DNA sequencing lies at the heart of the dramatic advances occurring in the life sciences. Computer graphics tools have proven essential for displaying and analyzing the enormous quantities of data and the complex patterns and relationships inherent in genetic analysis. Together, these capabilities are the major force behind today’s biotechnology revolution.
Acknowledgments
I would like to thank Cathy Frantz and her colleagues at Perkin-Elmer Applied Biosystems Division for providing much of the background material, including all the figures, for this article.
| Application areas
The development of DNA sequencing has led to an explosion of discoveries and capabilities. Areas profoundly affected include the following. Medical research. We have long known that certain diseases have genetic origins, such as sickle-cell anemia, cystic fibrosis, and hemophilia. In recent years, researchers have identified genes for many more diseases or susceptibilities, such as breast cancer, colon cancer, diabetes, Alzheimer's disease, even hereditary hearing loss, hypertension, and obesity. Identifying and sequencing the genes for a particular condition enables the development of diagnostics, or definitive tests for detecting the condition, as well as therapeutics, such as treatments, cures, or vaccines. Gene therapies can potentially fix or compensate for erroneous DNA sequences in diseased individuals. Genetic counseling can greatly aid potential partners in making reproductive choices. Genetic analysis is also a key in cancer and AIDS research. Basic life science research. This summer, scientists at the Institute for Genomic Research in Rockville, Maryland, sequenced the complete genome of archaea, regarded as the "third form of life." Archaea, discovered near volcanic jets on the ocean floor, can live in extreme temperatures and under enormous pressures. Sequencing revealed that only a third of archaea's DNA is the same as the other two more familiar forms of life, eukaryotes (all plants and animals whose cells have nuclei) and prokaryotes (bacteria), suggesting that it might be a distant ancestor to both. For these reasons, some consider archaea a candidate for life on other planets and the original life form on earth. Forensics. DNA analysis has taken on a large role in criminal investigations and trials such as the O.J. Simpson case. Tests for specific sets of DNA markers ("DNA fingerprinting") can show that suspects match blood, hair, semen, and other evidentiary samples with high probability, or definitively prove they don't match. The amplification properties of the polymerase chain reaction (PCR; see the section "Fragment sequencing") turn microscopic amounts of DNA into much larger quantities for testing. DNA techniques have also proven invaluable in identifying and reassembling the remains of victims in cases where fingerprints or dental records do not suffice. Animal husbandry. DNA analysis has helped verify the identity of highly valuable breeding animals such as livestock, racehorses, and purebred dogs. This can prove necessary when disputes, fraud, or "accidents" occur and in settling parentage, ownership, or insurance claims. Paternity disputes in humans are now frequently settled using the same techniques. Agriculture. DNA technology enables the development of crop and vegetable strains that are disease-, drought-, or spoil-resistant, higher yielding, or more nutritious. Hit-or-miss hybridization techniques used for years in crops such as corn have become much more sophisticated and effective using genetic engineering. Human Genome Project. This world-wide effort is tackling the ambitious task of sequencing the three-billion base-pair human genome in its entirety, with completion expected around the turn of the century. This undertaking rests on the ability of multiple laboratories around the world to consolidate and build on each others' efforts (see the Human Genome Project sites at http://www.ornl.gov/hgmis and http://www.er.doe.gov/production/oher/hug_top.html). Unlike other "big science" projects, requiring prohibitively expensive telescopes or particle accelerators, this project is accessible to any competent laboratory with a sequencing machine, computer, and connection to the Internet. This work's staggering potential includes helping detect and treat diseases, understanding development and reproduction, increasing longevity and quality of life, and even determining the origins of life. |
| Sizing the problem
Computer techniques are indispensable for attacking the DNA sequencing problem because of the enormous sequence lengths involved. Human DNA has more than three billion base pairs organized into 23 pairs of chromosomes present in every cell of the body (except red blood cells). Human chromosomes vary in length from 50 to 250 million base pairs. Scattered throughout the chromosomes are subsequences known as genes. These define the sets of proteins that determine each of the biological traits of an organism, such as blood type, susceptibility to particular diseases, production of key hormones like insulin, or characteristics like intelligence or physical appearance. Within the genes, adjacent groups of three base pairs, called codons, encode which of the 20 amino acids are used (plus start and stop points) when building a protein, in a process known as gene expression. Humans have approximately 100,000 genes, averaging 3,000 base pairs in length. Interestingly, the genes comprise only about two to three percent of the total human DNA. The rest consists of long repetitive sequences and other random patterns with no obvious function. Current sequencing technology can analyze DNA fragments of 500 or so base pairs. With three billion base pairs in human DNA, it takes six million such fragments to cover the entire length. Moreover, enough fragments with overlapping regions must be sequenced to reassemble the whole puzzle from these pieces. A few base pair changes at the right place in the genes determine an individual's specific characteristics, like eye color. About three million base pair choices make each of us (except for identical twins) genetically different. The remaining 99.9 percent of DNA is the same in all human beings. The sum of these DNA codes-the human genome-defines the human species. The DNA replication process is not exact, and errors, dropouts, or additions can occur. Single base-pair changes in the wrong place in a gene can result in nonviability of the organism or serious genetic diseases such as sickle-cell anemia. Yet errors in many other areas, including the 97 to 98 percent of the DNA with no apparent function, produce no obvious effects. |
| CG&A magazine page | Publications page | Computer Society home page |
Copyright (c) 1996 Institute of Electrical
and Electronics Engineers, Inc. All rights reserved.
Personal use of this material is permitted.
However, permission to reprint/republish this material for advertising
or promotional purposes or for creating new collective works for resale
or redistribution must be obtained from the IEEE. For information on obtaining
permission, send a blank email message to info.pub.permission@ieee.org
By choosing to view this document, you agree
to all provisions of the copyright laws protecting it.